
The past few years have seen a dramatic improvement in the capabilities of de novo protein design. This has extended more recently to de novo antibody design, as in just two years, the initial report of entirely de novo computationally-designed antibodies [1] has been followed by achievement of high “hit” rates and success against a broadening range of target classes by multiple groups, including at Xaira. Beyond simply increasing hit rates, de novo therapeutic design promises a more programmable form of discovery, including the ability to target specific epitopes, access to challenging targets and modalities that have historically been difficult to drug, and significant acceleration of early-stage discovery timelines.
As a vertically integrated company that combines frontier AI model development with therapeutic programs, we have experienced firsthand both the opportunities that de novo antibody design enables for therapeutic development and the challenges that come with translating those designs into therapeutic programs. Our active pipeline of de novo biologics across multiple indications (shown below) is a direct outcome of this approach and a key driver of our learning. As we advance this pipeline we continue to rigorously evaluate and benchmark our processes to ensure the molecules we design and select have the strongest possible potential to progress as developable candidates.
.jpg)
A recent and welcome note [2] raised the bar on two specific questions related to de novo designed molecules: is a reported binder real, and was its affinity measured in away the data can support? Their analysis of ambiguous sensorgrams, avidity artifacts from multivalent antigens, and over-fitted affinities provides an important cautionary note on how the field reports primary screening results, differentiating between initial “hits” and validated “binders”. In this post, we build on those considerations and aim to lay out our taxonomy around characterizing drug-like molecules as well as sharing with the field the bar we’ve set internally for a binder to become a viable therapeutic candidate. A molecule that genuinely binds is necessary, but binding-even well-measured, high-affinity binding-is fundamentally insufficient on its own.
Many molecules that bind a target with high affinity fail to translate into viable drug candidates. They may lack the right functional activity, exhibit poor biophysical properties, be immunogenic, or fall short of the requirements set in manufacturability and in vivo biology. Even binding measurements are only as reliable as the antigen used-poorly characterized or heterogeneous antigens can lead to misleading affinity, specificity, and epitope assessments, contributing to irreproducibility and false positives in early discovery.
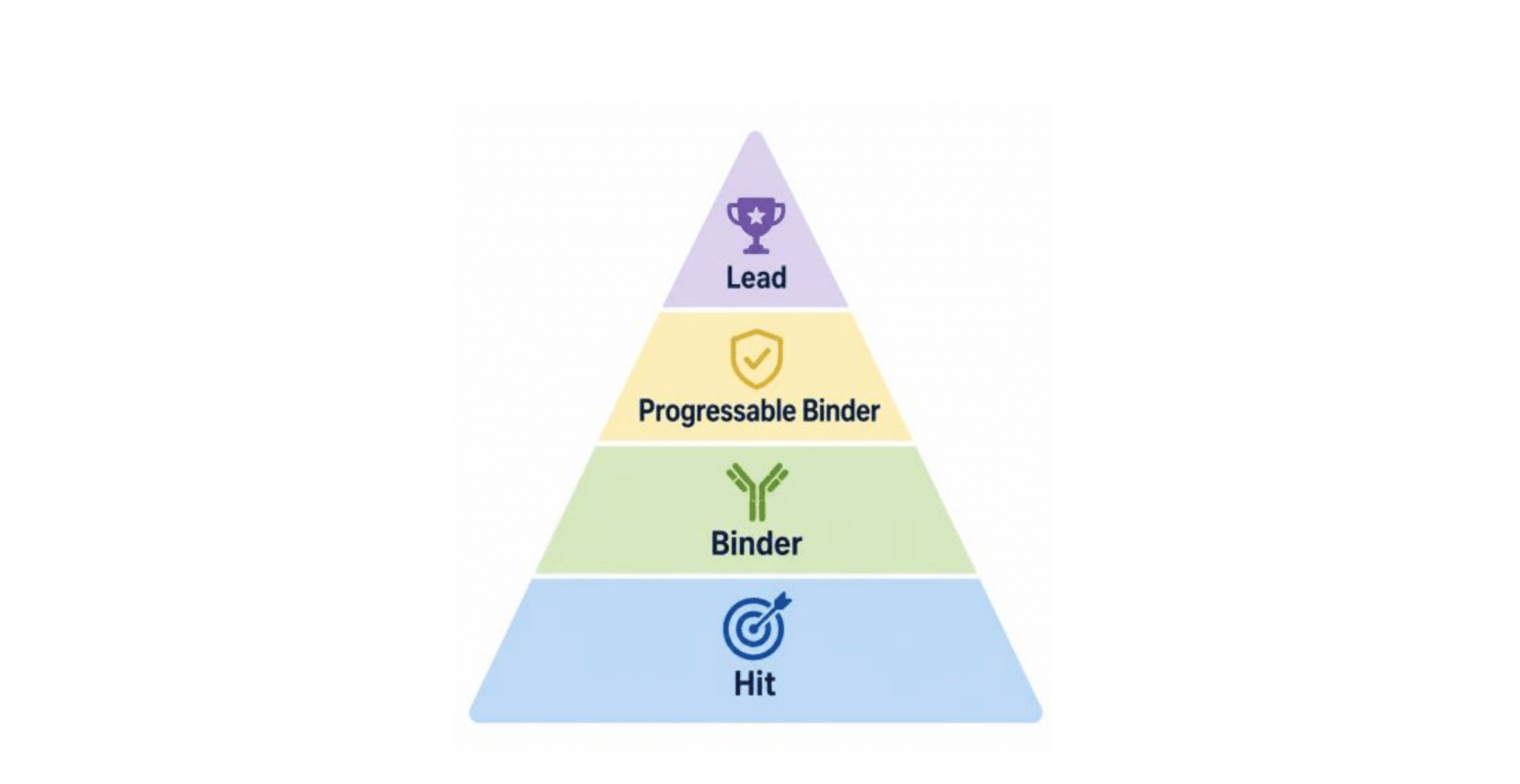
While ultimate success is only determined in the clinic, early-stage binders need to demonstrate a common set of characteristics that are measured in a rigorous and reproducible manner to successfully progress to later stages. To distinguish between other terms the AI design community have used, we refer to these as progressable binders - molecules that satisfy not just binding, but the broader set of functional, biophysical, and biological constraints required to advance into therapeutics. The historical basis for many of these constraints are well documented and practiced, having been identified as important, and in many cases critical, through more than 40 years of therapeutic antibody research: empirical biophysical boundaries derived from the clinical-stage antibody landscape [3], computational developability guidelines [4], and humanness/immunogenicity proxies validated against clinical data [5, 6].
The criteria are organized around three questions a candidate must answer in sequence:
- Is it a real binder? - antigen quality and rigorous binding measurement
- Is it drug-like? - developability, sequence integrity, immunogenicity, and canonical (CDR-mediated) binding
- Is it a therapeutic program? - functional activity and a diverse, de-risked panel of binders
In this post, we focus primarily on the first two questions, since they represent immediate challenges for the still nascent de novo design field, but we also highlight the importance of all three questions being answered to isolate development candidates to progress towards the clinic.
To be clear: these are enrichment filters, not clinical guarantees. We expand on their limits at the end.
Part 1 - Is it a real binder?
Here we extend the considerations in the recent post [2]
Soluble antigen prep and quality control (QC)
The quality of the soluble antigen ultimately defines the quality of the screen: without well-characterized reagents, even high-affinity binders can reflect measurement error rather than molecules that will translate into therapeutics. Therefore rigorous quality control of soluble antigens to ensure monodispersity, stability, and the absence of aggregation, as well as careful consideration of how constructs, tags, and immobilization strategies may alter epitope presentation is required.
For drug development, antigens should represent their native state as closely as possible, including correct folding, post-translational modifications, and oligomeric context. To best meet these criteria, as many drug targets are human proteins, it is common to utilize mammalian cells (i.e. HEK293) as an expression host as they can replicate correct folding and post-translational modifications of the antigen. E. coli and insect cell systems can produce many proteins, but lack or have different machinery for post-translational modifications. This may lead to non-representative antigen for drug discovery. Additionally, post-translational modifications and/or oligomeric states can be state dependent in a patient.
These considerations extend beyond biological relevance to how binding is measured in vitro: a target that dimerizes through an Fc tag, for example, can produce avidity-driven signals that inflate apparent affinity by orders of magnitude relative to the true monovalent interaction. Antigen preparation and format is therefore not a downstream detail - it is part of what determines whether a measured affinity is relevant.
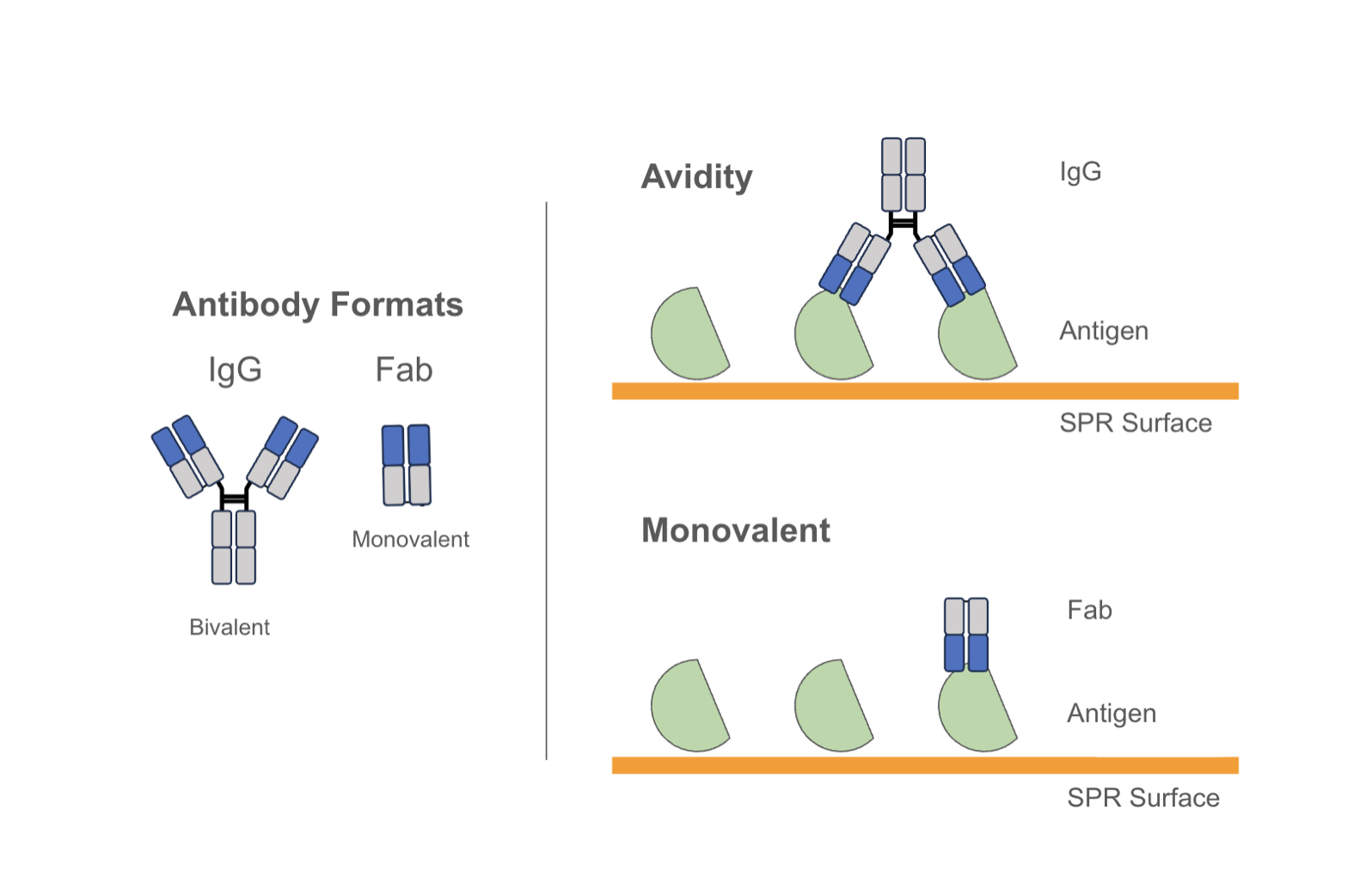
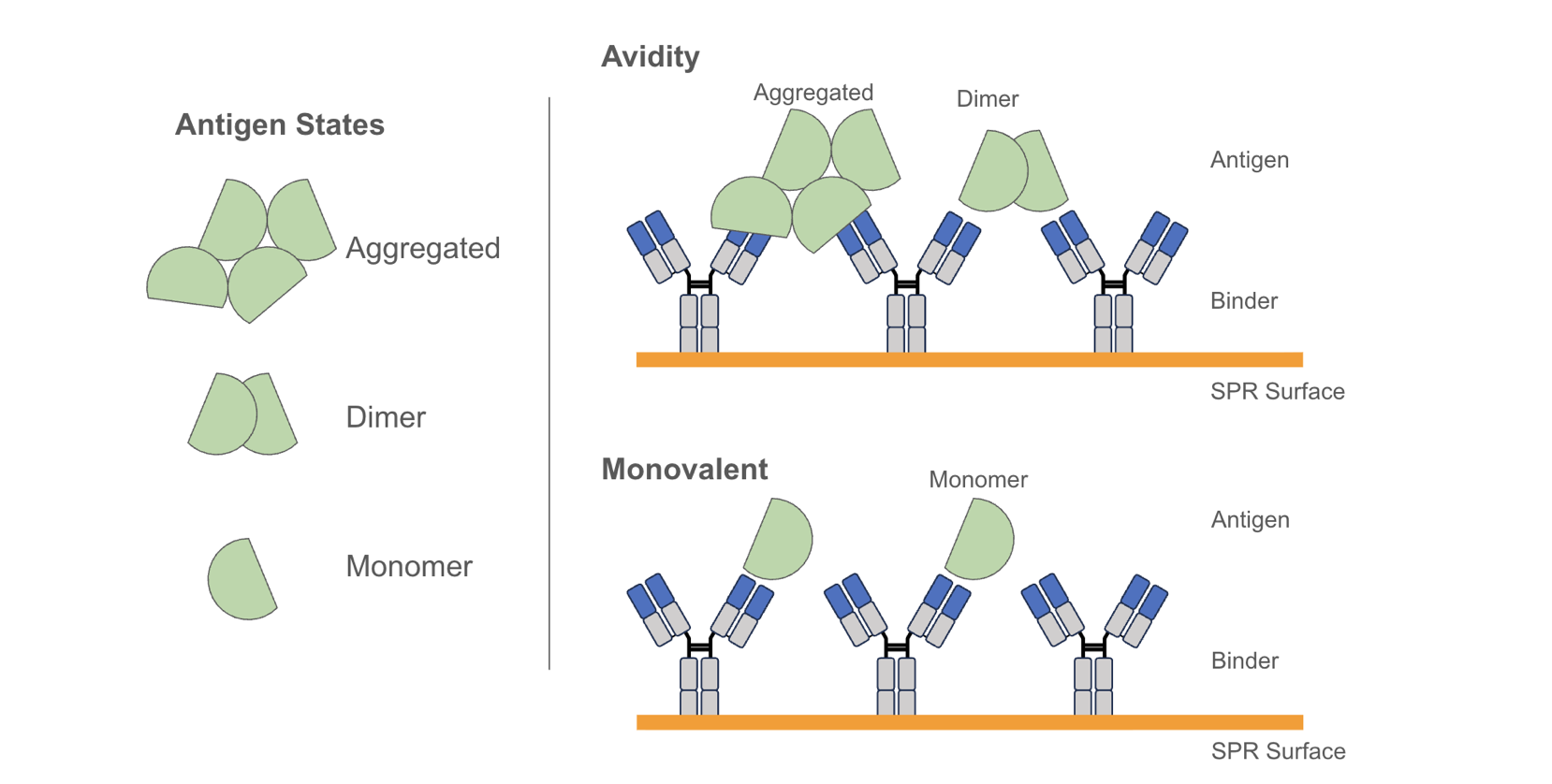
Membrane targets, especially multi-spanners such as GPCRs and ion channels, add another level of complexity to the process. “Soluble” forms of multi-spanner membrane targets (e.g. detergent soluble or nanodisc formats) are highly valuable, but not always technically feasible, or do not always retain functionally relevant activity. Therefore, the generation and use of target-expressing cell lines for multi-spanners is common. In the cases where cell lines are used, flow cytometry or cell-based ELISA assays can be employed to generate apparent Kd or cell-binding EC50 measurements. Again these measurements can be influenced by both the format of the binder and the expression level of the target on the cells, and are best reported as “cell-based Kds” or EC50s.
Measuring Binding
The affinity of an antibody to its target plays an important role in determining its efficacy. While equilibrium affinity (KD) is commonly reported as a summary metric, both on-rates and off-rates can individually impact the mechanism of action, as they govern target engagement and residence time in vivo.
Our antibody design model now regularly delivers initial hits (“zero-shot”) in the single digit or low double digit nanomolar, and even in the picomolar range (Figure 4A). But we also work with binders of lower affinity when seeking binders with specific properties against hard targets. Affinity maturation techniques can routinely improve binding from an initial hit, but the quality of the starting point still matters. Molecules with very weak initial affinity often require extensive optimization and are more likely to fail when additional constraints such as specificity, stability, and manufacturability are layered on. In our programs we therefore require early-stage binders to demonstrate an affinity of ~100 nM or tighter to the target. This threshold reflects a practical balance: it is permissive enough to allow discovery of diverse binders, while ensuring that candidates are within a regime where downstream optimization is minimized and tractable.
Crucially, that floor only means something if the underlying measurement is sound. We treat an affinity as real only when it derives from a confirmed binder - a clean sensorgram, or a signal corroborated by an orthogonal or flipped-orientation assay - and when the concentration series can support a fit. A reported "100 nM" hit that, on inversion of the assay or correction for target valency, resolves to micromolar monovalent affinity is not a 100 nM binder. We encourage reporting assay valency and releasing reference-subtracted sensorgrams so others can judge the signal for themselves. This is illustrated in Figure 4 with examples of de novo designs against COVID-RBD. Figures 4A and 4B show ~200 picomolar and double digit nanomolar zero-shot designs that represent validated binders. Figures 4C-4F illustrate molecules that failed validation in various ways.
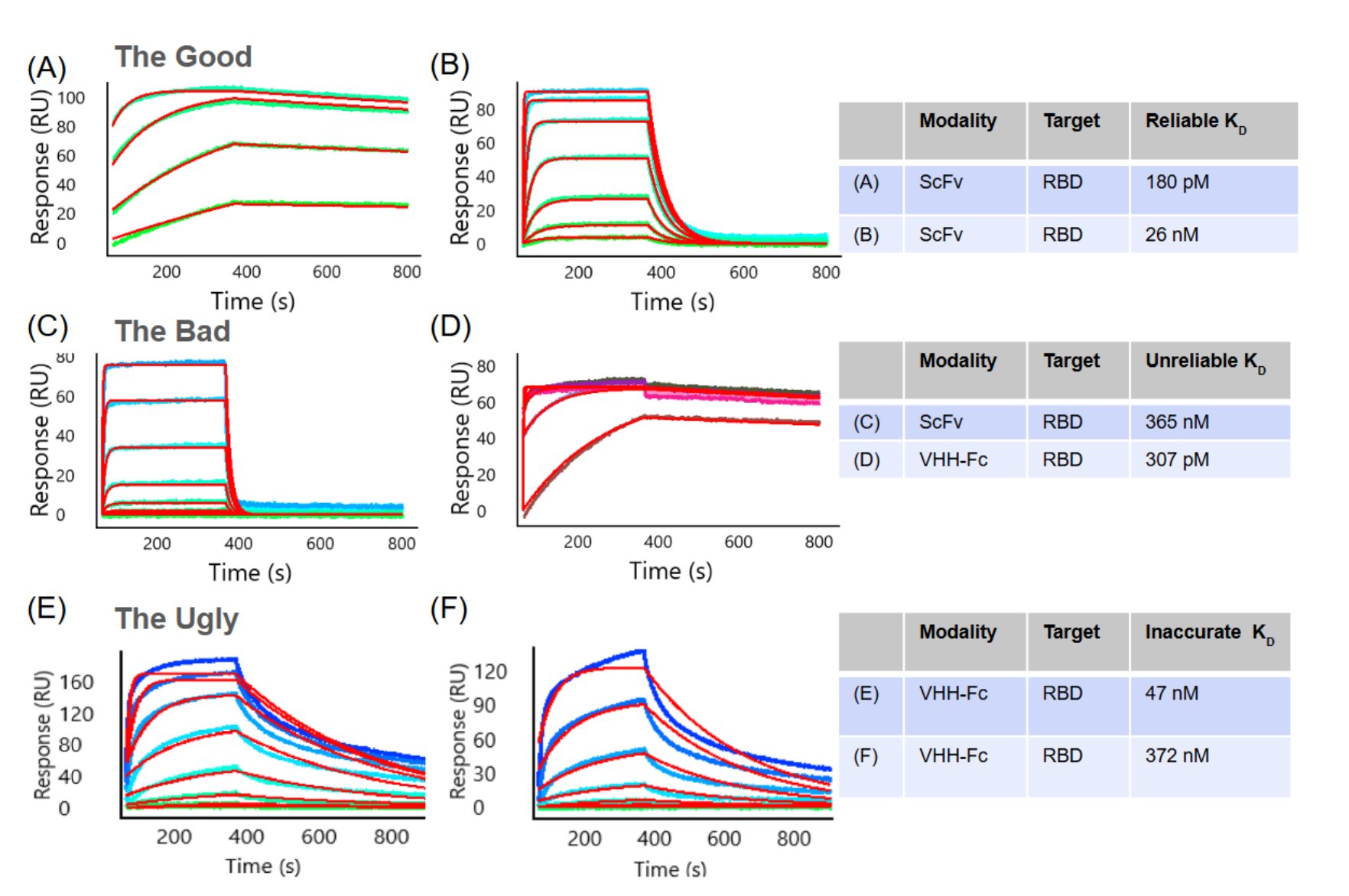
Figure 4: Examples of SPR sensorgrams from COVID-RBD studies.
In all panels, colored traces represent individual antigen concentrations and red lines indicate the global fit to the 1:1 Langmuir binding model. The assay set up is basically as shown in Figure 3B bottom right panel, with an anti-tag capture for scFvs, and an anti-Fc capture for VHH-Fcs, and varying concentrations of monovalent antigen as analyte.
Figure 4 A-B. The Good: Representative SPR sensorgrams demonstrating high-quality 1:1 Langmuir binding kinetics for antibody-antigen interactions at multiple antigen concentrations. (A) High-affinity interaction of a zero-shot de novo scFv design (KD = 180 pM) characterized by slow association and dissociation rates and good model fit. (B) Moderate affinity interaction of a different zero-shot scFv design (KD = 26 nM) with profiles enabling accurate equilibrium KD determination via steady-state analysis.
Figure 4 C-D. The Bad: Representative SPR sensorgrams illustrating common artifacts that compromise reliable KD determination in antibody-antigen binding measurements. (C) Fast-kinetics interaction where the antigen concentration range used does not reach saturation or Rmax, resulting in an unreliable software-generated KD of 365 nM. (D) Mass transport-limited interaction yielding an artificially low and unreliable software-generated KD of 307 pM. Note: for C and D the data quality issues can be addressed by refining the experimental parameters.
Figure 4 E-F. The Ugly: Representative SPR sensorgrams illustrating heterogeneous antibody-antigen interactions at multiple antigen concentrations. (E) Biphasic dissociation behavior is inconsistent with a simple 1:1 Langmuir binding model, as evidenced by the poor experimental curve and model fits (red lines). (F) In addition to biphasic dissociation, the progressive rise of the association curves without reaching saturation is characteristic of analyte aggregation. Note: for E and F underlying causes can include: (i) antibody binding to multiple distinct epitopes on the antigen; (ii) the presence of two or more antibody populations with differing orientations or conformations on the sensor surface; (iii) hydrophobic antibody regions promoting non-specific sticky interactions; (iv) antigen aggregation or dimerization leading to avidity effects; or (v) non-specific binding of the analyte to the sensor matrix, and can be harder to address.
Assessing success
In light of these considerations and those in [2], to enable cross-comparison of studies from different groups, we recommend reporting zero-shot success rates as the ratio of the number of validated binders (not initial hits) to the number of designs tested, with a clear enumeration of assay format (affinity, avidity, cell-based) and criteria for a hit counting as a binder (including cut-off, such as ~100 nM or better).
Part 2 - Is it drug-like?
Developability characteristics
Once binding is confirmed, the focus shifts to the biophysical properties, related to both biophysical and chemical stability, required for therapeutic application. Where applicable, we follow the empirical boundaries established for clinical-stage antibodies [3].
A progressable binder should have the following developability characteristics:
- Expressible in high-throughput mammalian systems with absolute yield sufficient for downstream characterization. Yield depends heavily on the production method; we require a minimum titer ≥15 µg/mL in our high throughput small scale TAP production. (Note: in assaying multiple designs in multiple formats to multiple antigens our titers typically range from 15 - 160 µg/mL with an average of ~ 50 µg/mL. Using slightly more time-consuming, plasmid-based production systems our typical titer is > 200 µg/mL. These levels are, of course, well below what is used for therapeutic production, but they are adequate for initial screening.)
- Thermally stable: first unfolding transition Tm1 ≥ 65°C for Fab formats and ≥ 60°C for VHH as measured by nanoDSF
- Monodisperse: ≥ 90% monomer content as assessed by SEC (single peak)
Specificity and selectivity are critical. To ensure low polyreactivity, we prioritize the following assays in this order:
- Meets the baculovirus particle (BVP) assay cutoff (< 3x assay background) [7]
- No detectable binding to non-target expressing cells
- Acceptable PAIATM assay characteristics (OVA, Heparin, Cation, and HIC)
Nonspecific binding is not merely an assay nuisance: polyreactivity is among the better-documented predictors of rapid clearance and PK failure, and avoiding it or engineering it out early is far cheaper than discovering it in vivo [8, 9]. Of course, binders that progress will undergo further polyspecificity and polyreactivity assays, and must also demonstrate requisite specificity over related or homologous epitopes.
Sequence liabilities
From a sequence perspective, we ensure designed antibodies are free of substantial liabilities [10], in particular:
- Preserve canonical disulfide bonds (C–C) within framework regions FR1–FR3 to ensure proper folding
- Introduce no additional cysteines.
- Carry no N-linked glycosylation motifs (NX(S/T)X) in their CDRs, where X does not equal P, to minimize heterogeneous glycosylation and interrupted binding events.
- Avoid known instability-prone motifs such as DP (acid-labile), DG (isomerization), and NG (deamidation).
Immunogenicity
Immunogenicity with subsequent development of anti-drug antibodies (ADAs), can have a major impact on therapeutic candidates. ADAs can reduce efficacy, alter pharmacokinetics, and in some cases drive adverse clinical responses. No preclinical assay fully predicts clinical immunogenicity, necessitating a conservative approach to managing this risk.
Mechanistically, immunogenicity is driven by peptide presentation on MHC molecules and subsequent activation of the adaptive immune response. As a result, the degree to which an antibody resembles human sequence and structural features can influence risk. This is especially relevant in VHH design, where native sequences are camelid-derived and typically require humanization. In early-stage discovery, in addition to leveraging in silico tools such as NetMHCIIpan [11], we prioritize binders that are substantially “human-like” by sequence-based metrics, e.g., OASis humanness scoring [5, 12] which has been shown to correlate with clinical immunogenicity. We treat these as imperfect but useful proxies for reducing immunogenicity risk, not as guarantees [as reviewed in 6]. In addition, as molecules progress through our pipeline they are subjected to a further panel of in vitro assays including MHC-Associated Peptide Proteomics (MAPPs) [13], T cell antigenicity [14] and dendritic cell presentation [15].
While each of these developability and immunogenicity properties is measured and reported separately, a progressable binder needs to simultaneously meet all these criteria - a higher bar. As an example, VHHs sampled from a naive camelid repertoire demonstrated a range of "developability" properties in our panel of assays (Figure 5). However, if humanness of the VHHs (as defined by the OASis scores [12]) is taken into account, the VHHs from the naive repertoire that are more human demonstrate greater developability challenges. This is not necessarily unexpected, as increasing humanness drives sequences away from those evolutionarily optimized in camelids, potentially disrupting framework features that support stability, solubility, and expression of native VHHs. Also of note, the pass rate for VHHs in the PAIA™️ hydrophobic interaction (HIC) assay (originally designed for classical antibodies) was generally low, consistent with recent data that suggests that the PAIA HIC-PLUS hydrophobicity assay is more relevant for VHH formats. This highlights the need to take the de novo molecule format into account when assessing developability to ensure the most relevant methodologies are used, and the importance of evaluating molecules simultaneously against all criteria as these properties can be in opposition to one another in individual designs.
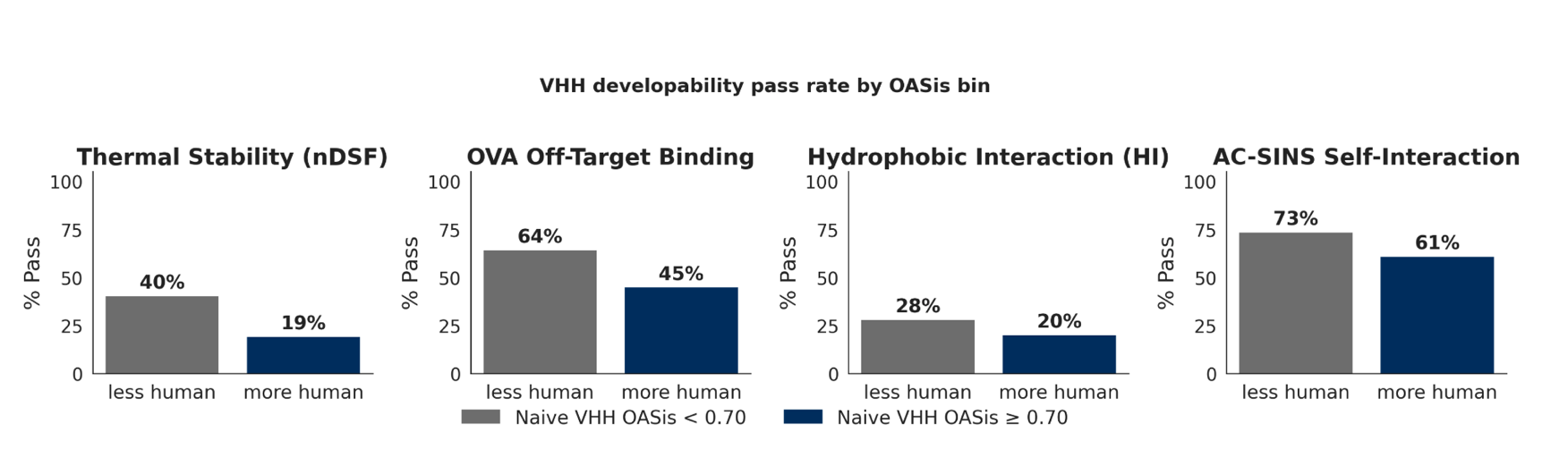
Distribution and pass rate of four developability assays for VHHs sourced from naive camelid repertoires (sequences post-aSEC purity filter and titer filter), stratified by OASis humanness score. Sequences predicted to be more human (OASis ≥ 0.7; the median clinical humanized-VHH threshold) measurably underperform their less-human counterparts on every developability property tested: thermal stability (Tm1 ≥ 65 °C: 41% → 20%), off-target binding by PAIA-OVA (OVA RFU ≥ 12: 64% → 46%), hydrophobic interaction by PAIA-HI (HI RFU ≥ 3: 28% → 19%), and self-interaction (AC-SINS shift ≤ 5 nm: 74% → 61%). Thus while sampling sequences close to the camelid distribution might naturally result in developable sequences, progressable binders need to be human and developable - a higher bar.
CDR-Mediated Interactions
Antibodies generated by the immune system acquire antigen binding through V(D)J recombination followed by somatic hypermutation and affinity maturation, a process that strongly favors CDR-mediated antigen recognition. In contrast, de novo antibody design is not constrained by the same evolutionary and selection pressures. As a result, generative models may produce antibodies whose predicted antigen contacts extend beyond the canonical CDR paratope and involve framework residues. These binding modes are not inherently non-functional, but they are non-canonical and may require additional scrutiny because they could be less robust, more difficult to optimize, or present greater developability risk. For this reason, de novo-designed antibodies should be explicitly evaluated for framework-mediated antigen contacts and other atypical binding architectures.
Several approaches have addressed this by incorporating structural or sequence-based constraints into training or scoring functions, encouraging paratope localization within the CDRs. This issue can be particularly relevant in VHH design, where the reduced scaffold can make the balance of framework versus CDR contributions more variable.
In our programs, we therefore require that a progressable design be predicted to engage the antigen predominantly through CDR-mediated interactions. For example, the design shown in the right of figure below is predicted (computationally) to bind to the PDL1 antigen via significant framework contacts. This design would not pass our criteria and would be filtered out, in contrast to the design on the left that would pass our filters.
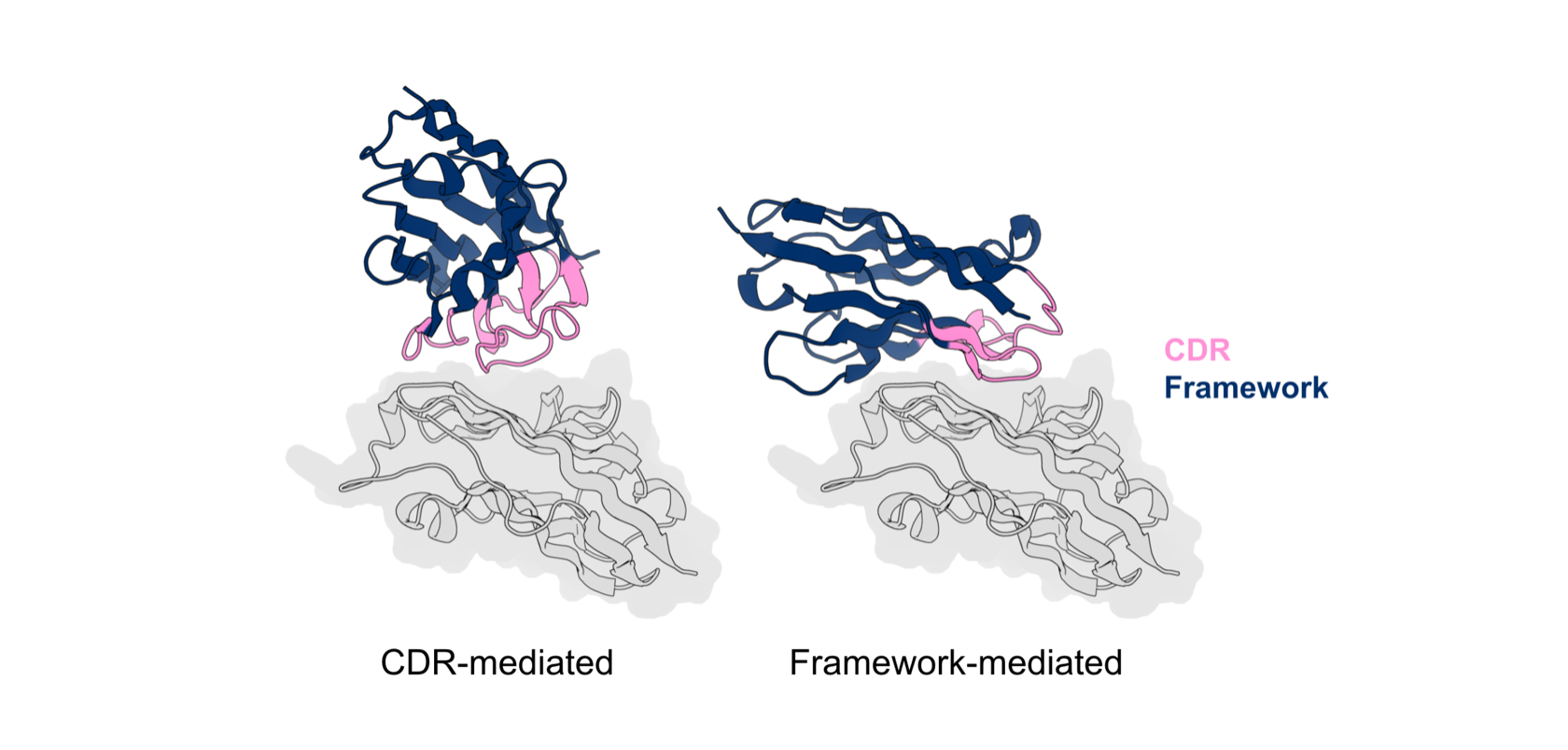
In silico models of designed VHH interacting with target antigen (PDL1). The VHH on the left primarily interacts with the antigen through the designed CDR while the VHH on the right interacts with the antigen through the native framework and the designed CDR.
Assessing success
While many groups report distributions of individual properties across designed binders, these summaries do not capture whether any single design satisfies all requirements necessary for progression. Instead, we recommend reporting zero-shot progressable rates as the fraction of designs that simultaneously meet all defined criteria, rather than highlighting property-wise distributions in isolation with a clear enumeration of diversity thresholds and filters for CDR mediated interactions.
Part 3 - Is it a therapeutic program?
Functional characterization
Binding and developability establish that a molecule could be a drug; functional activity establishes that it may have a therapeutic effect. Because the mechanism of action is target- and modality-specific, we define functionality on a target-by-target basis rather than against a universal assay and/or cutoff. Depending on the program, this may include agonism or antagonism in a cell-based reporter or signaling assay, blockade of a ligand–receptor interaction, internalization and trafficking for payload-bearing formats, or effector function (e.g., ADCC/CDC) where engagement of immune mechanisms is intended.
Diversity of Binders
Meeting each criterion above improves the probability of downstream success for any given molecule, but individual molecules still fail at later stages for functional, biophysical, or developability reasons. Routine program success therefore depends not only on identifying progressable binders, but on being able to identify a sufficiently diverse set of such candidates. In an example, having 10 progressable binders that only differ by one amino acid between them is not nearly as powerful of a diversification strategy as having 10 progressable binders that each bring a unique element of de-risking for the many downstream requirements awaiting them.
We require our internal programs to enter lead optimization only when we have obtained a structurally and functionally diverse panel of progressable binders. In practice, this diversity spans multiple axes, including but not limited to, targeted epitope, binding geometry, CDR sequence, and structure. Maintaining diversity across these dimensions increases the likelihood that at least one molecule exhibits a favorable balance of potency, specificity, and developability, while reducing the risk of over-committing to a single molecule early in the program. This emphasis on diversity, in turn, necessitates designing and evaluating a larger initial set of candidates than hit rates alone would suggest, since a high hit rate could result from overrepresentation of a single cluster of closely related designs.
A note on limits:
These criteria are enrichment filters, not a clinical oracle. Thresholds are modality - and target-dependent, and even a fully conforming panel carries program risk; conversely, the clinical-stage landscape includes approved antibodies that would flag on one or more biophysical metrics. This bar substantially enriches molecules for potential success - but doesn’t guarantee them. We share some of our criteria for de novo AI-designed molecules in that spirit, and welcome scrutiny and counterexamples from the community to move the field forward together for the benefit of patients.
Summary: The Progressable Binder Standard
De novo antibody design has made significant progress with respect to hit rates, but we believe the problem requires a more holistic focus on therapeutic development. Towards this, we introduce our definition of a “progressable binder,” one that encompasses both extrinsic (high affinity, low predicted immunogenicity, functional efficacy) as well as intrinsic (favorable developability) properties to maximize chances of (pre-)clinical success. Finally, we believe maximizing therapeutic translation towards clinical success requires moving away from the mindset of a “single best hit” toward a structurally and functionally diverse panel of candidates equipped to survive downstream pre-clinical and clinical challenges.
[[divider]]
Acknowledgements: thank you to everyone at Xaira who made this work possible. Special thanks to Andy Deng, Anna Lauko, Do Soon Kim, Tilelii Amimeur and Zimple Matharu for input on this document.
References:
- Bennett, N. R., Watson, J. L., Ragotte, R. J., Borst, A. J., See, D. L., Weidle, C., Biswas, R., Yu, Y., Shrock, E. L., Ault, R., Leung, P. J. Y., Huang, B., Goreshnik, I., Tam, J., Carr, K. D., Singer, B., Criswell, C., Wicky, B. I. M., Vafeados, D., Sanchez, M. G., Kim, H. M., Torres, S. V., Chan, S., Sun, S. M., Spear, T., Sun, Y., O'Reilly, K., Maris, J. M., Sgourakis, N. G., Melnyk, R. A., Liu, C. C., & Baker, D. (2025). Atomically accurate de novo design of antibodies with RFdiffusion. bioRxiv 2024.03.14.585103. https://www.biorxiv.org/content/10.1101/2024.03.14.585103v2
- Boltz. (2026). Experimental protocol. https://boltz.bio/experimental-protocol
- Jain, T., Sun, T., Durand, S., Hall, A., Houston, N. R., Nett, J. H., Sharkey, B., Bobrowicz, B., Caffry, I., Yu, Y., Cao, Y., Lynaugh, H., Brown, M., Baruah, H., Gray, L. T., Krauland, E. M., Xu, Y., Vásquez, M., & Wittrup, K. D. (2017). Biophysical properties of the clinical-stage antibody landscape. Proceedings of the National Academy of Sciences, 114(5), 944–949. https://doi.org/10.1073/pnas.1616408114
- Raybould, M. I. J., Marks, C., Krawczyk, K., Taddese, B., Nowak, J., Lewis, A. P., Bujotzek, A., Shi, J., & Deane, C. M. (2019). Five computational developability guidelines for therapeutic antibody profiling. Proceedings of the National Academy of Sciences, 116(10), 4025–4030. https://doi.org/10.1073/pnas.1810576116
- Prihoda, D., Maamary, J., Waight, A., Juan, V., Fayadat-Dilman, L., Svozil, D., & Bitton, D. A. (2022). BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. mAbs, 14(1), 2020203. https://doi.org/10.1080/19420862.2021.2020203
- Agnihotri, S., Gonzalez-Nolasco B., Monian, B., Pattijn, S., Ackaert, C., Wu, P., Kettenberger, H., Tourdot, S., Hickling, T., Hu, Z., Higgs, R. E., & Leventhal, D. S. (2025). The Immunogenicity Database Collaborative (IDC): A standardized, publicly available database for clinical immunogenicity observations and insights. bioRxiv 2025.12.08. https://doi.org/10.64898/2025.12.08.692993
- Hötzel, I., Theil, F.P., Bernstein, L.J., Prabhu, S., Deng, R., Quintana, L., Lutman, J., Sibia, R., Chan, P., Bumbaca, D., Fielder, P., Carter, P.J., & Kelley, R.F. (2012). A strategy for risk mitigation of antibodies with fast clearance. mAbs, 4(6), 753–760. https://doi.org/10.4161/mabs.22189
- Cunningham, O., Scott, M., Zhou, Z. S., & Finlay, W. J. J. (2021). Polyreactivity and polyspecificity in therapeutic antibody development: Risk factors for failure in preclinical and clinical development campaigns. MAbs, 13(1), 1999195. https://doi.org/10.1080/19420862.2021.1999195
- Starr, G. S., & Tessier, P. M. (2019). Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotechnology, 26(60), 119-127. https://doi.org/10.1016/j.copbio.2019.01.008
- Teixeira, A.A.R., D'Angelo, S., Erasmus, M.F., Leal-Lopes, C., Ferrara, F., Spector, L.P., Naranjo, L., Molina, E., Max, T., DeAguero, A., Perea, K., Stewart, S., Buonpane, R.A., Nastri, H.G., Bradbury, A.R.M. (2022). Simultaneous affinity maturation and developability enhancement using natural liability-free CDRs. mAbs, 14(1), 2115200. https://doi.org/10.1080/19420862.2022.2115200
- Reynisson, B., Alvarez, B., Pau,l S., Peters, B., & Nielsen, M. (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48(W1):W449-W454. https://doi.org/10.1093/nar/gkaa379
- Kovaltsuk, A., Leem, J., Kelm, S., Snowden, J., Deane, C.M., & Krawczyk, K. (2018). Observed Antibody Space: A Resource for Data Mining Next-Generation Sequencing of Antibody Repertoires. J Immunol. 201(8), 2502-2509. https://doi.org/10.4049/jimmunol.1800708
- Sekiguchi, N., Kubo, C., Takahashi, A., Muraoka, K., Takeiri, A., Ito, S., Yano, M., Mimoto, F., Maeda, A., Iwayanagi, Y., Wakabayashi, T., Takata, S., Murao, N., Chiba, S., & Ishigai, M. (2018). MHC-associated peptide proteomics enabling highly sensitive detection of immunogenic sequences for the development of therapeutic antibodies with low immunogenicity. mAbs. 10(8), 1168-1181. https://doi.org/10.1080/19420862.2018.1518888
- Cohen, S., Myneni, S., Batt, A., Guerrero, J., Brumm, J., & Chung, S. (2021). Immunogenicity risk assessment for biotherapeutics through in vitro detection of CD134 and CD137 on T helper cells. mAbs. 13(1), 1898831. https://doi.org/10.1080/19420862.2021.1898831
- Ackaert, C., Gonzalez-Nolasco, B., Rosenbaum, M., Perez-Olivares, M., Gutknecht, M., Ducret, A., & Karle, A.C. (2025). Dendritic cell maturation assay for non-clinical immunogenicity risk assessment: best practices recommended by the European Immunogenicity Platform. Front Immunol. 16:1704045. https://doi.org/10.3389/fimmu.2025.1704045